GROMACS Tutorials
Justin A. Lemkul, Ph.D.
Associate Professor
Virginia Tech Department of Biochemistry
All tutorials are compatible with GROMACS version 2018 and are being updated for version 2025!
FUNDAMENTALS
Tutorial 1: Lysozyme in Water |
|
Tutorial 3: Protein-Ligand Complex |
ADVANCED TOPICS
Tutorial 4: van der Waals Transformation |
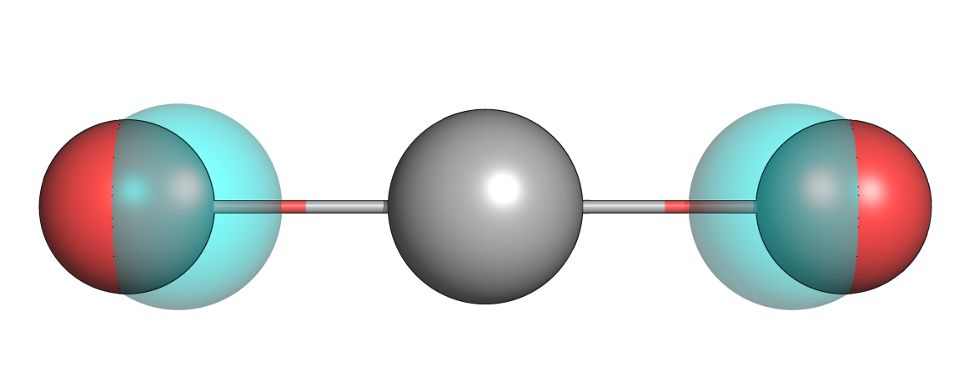
Tutorial 5: Ethanol Hydration Free Energy |
Coming soon: Protein-Ligand Binding Free Energy |

|
|
|
|
These tutorials are designed as introductory material into using the GROMACS simulation package. GROMACS is free, open-source software, and has consistently been one of the fastest (if not the fastest) molecular dynamics codes available. There are currently eight tutorials available, with a ninth in the works:
All of these tutorials assume you are using GROMACS version 2018 or newer. Some (as noted in the tutorials themselves) require version 2025. If you are using a different version, be forewarned: the tutorials likely will not work as expected. At the end of each tutorial you will find my contact information if you wish to provide commentary or report anything you find to be incorrect. I genuinely appreciate this kind of feedback, as it helps me design better tutorials and fix things that are not clear (or sometimes wrong, oops). I must ask that you please do not contact me for general GROMACS help or advice on your project. I am continually inundated with help requests and I simply do not have the time to be helpful to everyone. I hope you find these tutorials useful. If you use these protocols for your research, I ask that you cite the paper that explains the theoretical background for the procedures: J.A. Lemkul (2018) "From Proteins to Perturbed Hamiltonians: A Suite of Tutorials for the GROMACS-2018 Molecular Simulation Package, v1.0" Living J. Comp. Mol. Sci. 1 (1): 5068. Happy simulating! For GROMACS installation instructions, refer here. |
Site design and content copyright Justin Lemkul
The contents of all tutorials on this site are made available under the CC-BY 4.0 license
Problems with the site? Send them to the Webmaster