GROMACS Tutorial
Step Nine: Analysis
Analyzing Compactness: RgThe radius of gyration of a protein is a measure of its compactness. If a protein is stably folded, it will likely maintain a relatively steady value of Rg. If a protein unfolds, its Rg will change over time. Let's analyze the radius of gyration for lysozyme in our simulation: gmx gyrate -s md_0_10.tpr -f md_0_10_noPBC.xtc -o gyrate.xvg -sel Protein -tu ns 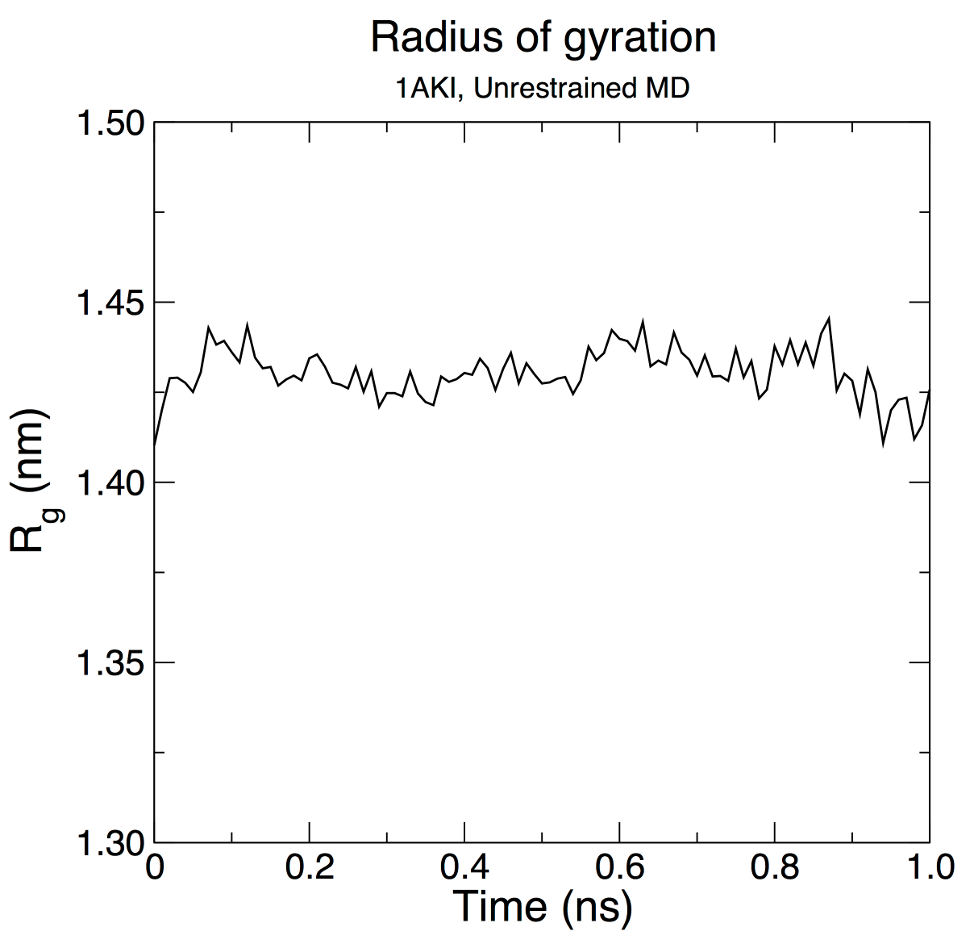
We can see from the reasonably invariant Rg values (1.409 ± 0.008 nm) that the protein does not undergo any substantial elongation or compaction relative to its starting structure. Thus, it remains in its compact (folded) form over the course of 10 ns at 298 K. This result is not unexpected, but illustrates an advanced capacity of GROMACS analysis that comes built-in. As with the RMSD plot on the previous page, the data have been smoothed over 500-ps running windows in XmGrace. Secondary StructureProtein structure is often assessed in terms of secondary structure, that is, the persistence of α-helices, β-sheets, etc. The GROMACS program gmx dssp invokes the Dictionary of Secondary Structure of Proteins (DSSP) algorithm to assign secondary structure to each residue in the protein. To run this analysis, execute the following command: gmx dssp -s md_0_10.tpr -f md_0_10_noPBC.xtc -tu ns -o dssp.dat -num dssp_num.xvg After making some adjustments to labeling (there are misformatted characters in the output of version 2025.2) and reordering the data series, one can obtain a plot similar to the following in XmGrace from dssp_num.xvg. The solid lines again correspond to a 500-ps (50-frame) running average to smooth fluctuations. 
Hydrogen BondsGROMACS uses two criteria to determine if a hydrogen bond exists between two groups: (1) the donor-H distance (defaults to 0.35 nm or less, can be tuned with -hbr) and (2) the donor-acceptor-H angle (defaults to 30°, can be tuned with -hba). Note the order of atoms in the angle criterion; GROMACS does not use the donor-H-acceptor angle as is employed in other software. Hydrogen bonds are highly directional and are strongest when linear, so the use of donor-acceptor-H ≤ 30° means that donor-H-acceptor angles will range from 150° - 180°. The user is prompted for two selections when computing hydrogen bonds. These groups must be either identical or completely distinct (no shared atoms). Several examples are illustrated below. The first is the number of hydrogen bonds within the backbone of the protein. gmx hbond -s md_0_10.tpr -f md_0_10_noPBC.xtc -tu ns -num hbnum_mainchain.xvg Select the "MainChain+H" group (7) for both selections when prompted. In GROMACS, the "MainChain" group contains the N, Cα, C, and O atoms. "MainChain+H" includes those four atoms, plus the amide H atom, which is required for assignment of hydrogen bonds. In GROMACS convention, "Backbone" contains only N, Cα, and C atoms; no hydrogen bonds can be computed from this group, despite the vernacular of "backbone hydrogen bonds." Next, compute hydrogen bonds formed among sidechain atoms: gmx hbond -s md_0_10.tpr -f md_0_10_noPBC.xtc -tu ns -num hbnum_sidechain.xvg Select the "SideChain" group (8) for both selections. Finally, we will compute hydrogen bonds between the protein and water: gmx hbond -s md_0_10.tpr -f md_0_10_noPBC.xtc -tu ns -num hbnum_prot_wat.xvg Select "Protein" (1) and "Water" (12) or "SOL" (13) as the two groups ("Water" and "SOL" are equivalent). Plotted together, the results should resemble what is shown below: 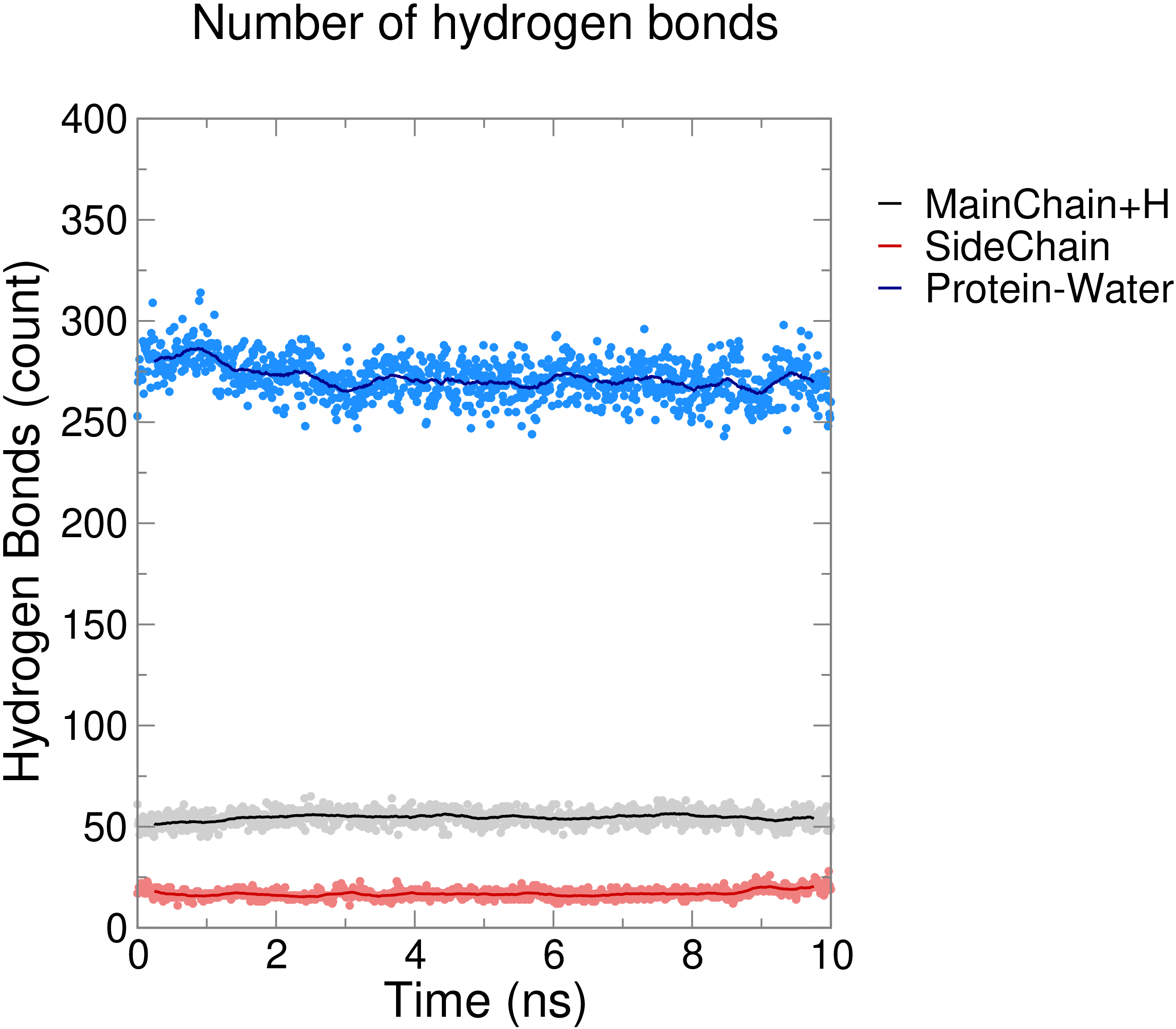
In each case, the number of hydrogen bonds is relatively consistent over time, which is to be expected in the case of a short simulation like this one. Lysozyme forms a large number of hydrogen bonds with water, roughly 55 within the "backbone" (MainChain+H), and fewer (~20) among sidechain atoms. |
Site design and content copyright Justin Lemkul
Problems with the site? Send them to the Webmaster