GROMACS Tutorial
Step Five: Energy Minimization
|
The solvated, electroneutral system is now assembled. Before we can begin dynamics, we must ensure that the system has no steric clashes or inappropriate geometry. The structure is relaxed through a process called energy minimization (EM). The process for EM is much like the addition of ions. We are once again going to use grompp to assemble the structure, topology, and simulation parameters into a binary input file (.tpr), but this time, instead of passing the .tpr to genion, we will run the energy minimization through the GROMACS MD engine, mdrun. Assemble the binary input using grompp using this input parameter file: gmx grompp -f inputs/minim.mdp -c 1AKI_solv_ions.gro -p topol.top -o em.tpr Make sure you have been updating your topol.top file when running genbox and genion, or else you will get lots of nasty error messages ("number of coordinates in coordinate file does not match topology," etc). We are now ready to invoke mdrun to carry out the EM: gmx mdrun -v -deffnm em The -v flag is for the impatient: it makes mdrun verbose, such that it prints its progress to the screen at every step. The -deffnm flag will define the file names of the input and output. So, if you did not name your grompp output "em.tpr," you will have to explicitly specify its name with the mdrun -s flag. In our case, we will get the following files:
The mdrun program will also print the final output of energy minimization (maximum force, and the potential energy of the system) to both the .log file and the terminal. You should see something like this in the terminal: Steepest Descents converged to Fmax < 1000 in 566 steps Potential Energy = -6.2751356e+05 Maximum force = 9.8003864e+02 on atom 567 Norm of force = 2.3013127e+01 There are two very important factors to evaluate to determine if EM was successful. The first is the potential energy (printed at the end of the EM process, even without -v). Epot should be negative, and (for a simple protein in water) on the order of 105-106, depending on the system size and number of water molecules. The second important feature is the maximum force, Fmax, the target for which was set in minim.mdp - "emtol = 1000.0" - indicating a target Fmax of no greater than 1000 kJ mol-1 nm-1. It is possible to arrive at a reasonable Epot with Fmax > emtol. If this happens, your system may not be stable enough for simulation. Evaluate why it may be happening, and perhaps change your minimization parameters (integrator, emstep, etc). Let's do a bit of analysis. The em.edr file contains all of the energy terms that GROMACS collects during EM. You can analyze any .edr file using the GROMACS energy module: gmx energy -f em.edr -o potential.xvg At the prompt, type "11 0" to select Potential (11); zero (0) terminates input. You will be shown the average of Epot, and a file called "potential.xvg" will be written. To plot this data, you will need the Xmgrace plotting tool. The resulting plot should look something like this, demonstrating the nice, steady convergence of Epot: 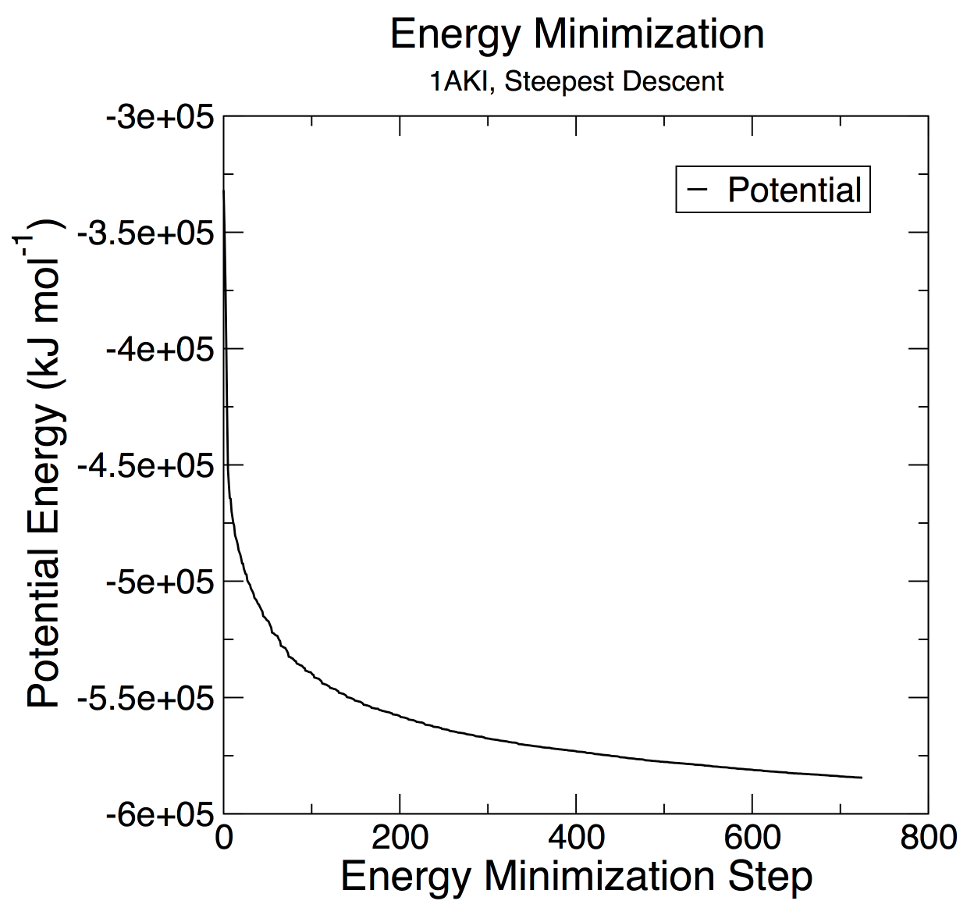
Now that our system is at an energy minimum, we can begin real dynamics. |
Site design and content copyright Justin Lemkul
Problems with the site? Send them to the Webmaster