GROMACS Tutorial
Final Notes: Tips & Tricks
|
Adding a Protein to the Aqueous Layer Interested in how a protein or peptide might interact with such a biphasic system? The same principles as before apply to positioning the protein: place the protein in the unit cell of desired dimensions, and manually set its center. For our system, we can assign a generic peptide to a position in the "top" half of the box with editconf like so: gmx editconf -f peptide.gro -o peptide_newbox.gro -box 4.30795 4.30795 8.6159 -center 2.153975 2.153975 6.461925 The assigned z-position for the protein center is three-quarters of the box length (original cyclohexane box plus one-half). How do we place that protein in the same unit cell as our cyclohexane layer? We could use the Unix comand gmx solvate -cp peptide_newbox.gro -cs chx_newbox.gro -o peptide_chx.gro Using the KALP15 peptide from the membrane protein tutorial, the system would look something like this: 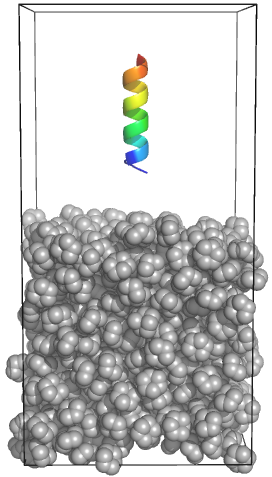
Then proceed with another round of solvate to add water, as before. Energy-minimize, equilibrate, and simulate. Expanding the Box Let's say your protein is too large for a box that is only ~4.3 nm in the x-y plane. It is possible to expand your system simply by using genconf: gmx genconf -f chx_10ns.gro -nbox 2 2 2 -o chx_bigbox.gro The above command will create a new, 8.6-nm cube of cyclohexane with 8 times the number of molecules in it. Again, it is not necessary to create a cube. Then follow all the steps outlined previously for positioning this new, larger layer in a box, adding whatever protein or solute you desire, and filling the box with water. In lieu of a new, cubic box, an expanded layer can be created along the x-y plane with a slightly different command: gmx genconf -f chx_10ns.gro -nbox 2 2 1 -o chx_biglayer.gro Summary You have now built a biphasic cyclohexane-water system, and hopefully understand the basic principles of how to place any arbitrary molecule into such a system in a desired location. If you have suggestions for improving this tutorial, if you notice a mistake, or if anything is otherwise unclear, please feel free to email me. Please note: this is not an invitation to email me for GROMACS problems. I do not advertise myself as a private tutor or personal help service. That's what the GROMACS User Forum is for. I may help you there, but only in the context of providing service to the community as a whole, not just the end user. Happy simulating!
|
Site design and content copyright Justin Lemkul
Problems with the site? Send them to the Webmaster