GROMACS Tutorial
Step Five: Generating Configurations
|
To conduct umbrella sampling, one must generate a series of configurations along a reaction coordinate, ζ. Some of these configurations will serve as the starting configurations for the umbrella sampling windows, which are run in independent simulations. The figure below illustrates these principles. The top image illustrates the pulling simulation we will run now, conducted in order to generate a series of configurations along the reaction coordinate. These configurations are extracted after the simulation is complete (dashed arrows in between the top and middle images). The middle image corresponds to the independent simulations conducted within each sampling window, with the center of mass of the free peptide restrained in that window by an umbrella biasing potential. The bottom images shows the ideal result as a histogram of configurations, with neighboring windows overlapping such that a continuous energy function can later be derived from these simulations. 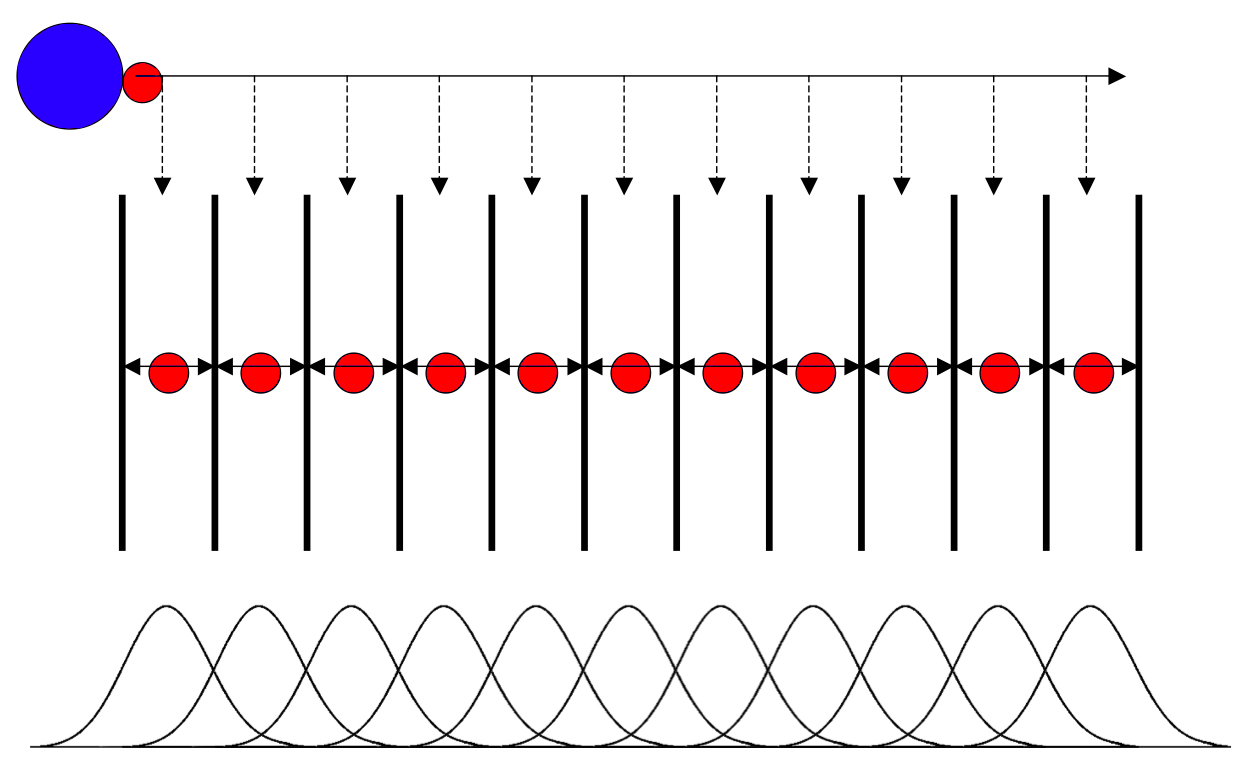
For this example, the reaction coordinate is the z-axis. To generate these configurations, we must pull peptide A away from the protofibril. We will pull over the course of 500 ps of MD, saving snapshots every 1 ps. This setup has been established based on trial-and-error to obtain a reasonable distribution of configurations. In other systems, it may be necessary to save configurations more often, or sufficient to save configurations less often. The idea is to capture enough configurations along the reaction coordinate to obtain regular spacing of the umbrella sampling windows, in terms of center-of-mass distance between peptides A and B. The .mdp file for this pulling can be found here. A brief explanation of the pulling options used is as follows: ; Pull code pull = yes pull_ncoords = 1 ; only one reaction coordinate pull_ngroups = 2 ; two groups defining one reaction coordinate pull_group1_name = Chain_A pull_group2_name = Chain_B pull_coord1_type = umbrella ; harmonic potential pull_coord1_geometry = distance ; simple distance increase pull_coord1_dim = N N Y ; pull along z pull_coord1_groups = 1 2 ; groups 1 (Chain A) and 2 (Chain B) define the reaction coordinate pull_coord1_start = yes ; define initial COM distance > 0 pull_coord1_rate = 0.01 ; 0.01 nm per ps = 10 nm per ns pull_coord1_k = 1000 ; kJ mol^-1 nm^-2
Remember that We will need to define some custom index groups for this pulling simulation. Use make_ndx: gmx make_ndx -f npt.gro ... > r 1-27 > name 19 Chain_A > r 28-54 > name 20 Chain_B > q Now, run the steered MD simulation: gmx grompp -f md_pull.mdp -c npt.gro -p topol.top -r npt.gro -n index.ndx -t npt.cpt -o pull.tpr gmx mdrun -deffnm pull -pf pullf.xvg -px pullx.xvg The process will play out something like the following: 
There is no displacement for some time, as the force on the imaginary spring builds up until it is sufficient to overcome the restoring forces within the protofibril structure. About halfway through the SMD simulation, chain A begins to dissociate from chain B. This process is also visible in the force on the spring over time, stored in pullf.xvg: 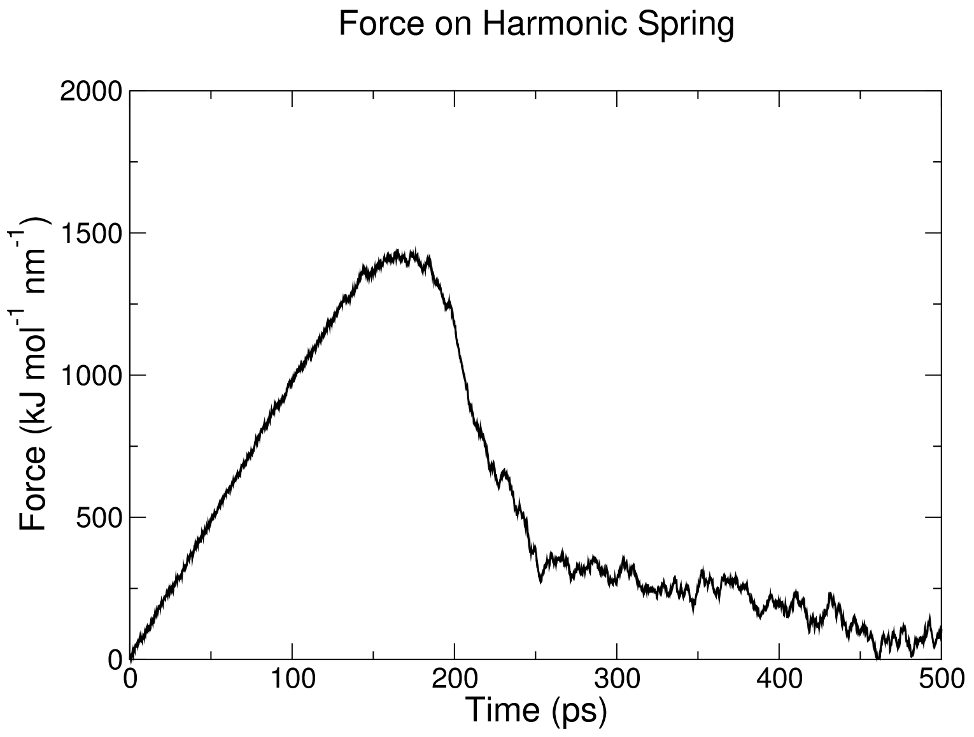
To prepare the individual umbrella sampling windows, we will need to extract useful frames from the SMD trajectory. The easiest way I have found to do this is the following:
To extract the frames from your trajectory (traj.xtc), use trjconv (save the whole system, group 0, when prompted): gmx trjconv -s pull.tpr -f pull.xtc -o conf.gro -sep A series of coordinate files (conf0.gro, conf1.gro, etc) will be produced, corresponding to each of the frames saved in the continuous pulling simulation. To iteratively call gmx distance on all of these (501!) frames that were generated, I have written a Bash script that takes care of this task. It will print a file called "summary_distances.dat" that contains this information. The script can be found here. Look at the contents of summary_distances.dat to see the progression of COM distance between chain A and chain B over time. Make note of the configurations to be used for umbrella sampling, based on the desired spacing. That is, if you want 0.2-nm spacing, you might find the following lines in summary_distances.dat: 6 0.500 ... 160 0.704 You would then use conf6.gro and conf160.gro as the starting configurations of two adjacent umbrella sampling windows. Make note of all the configurations you wish to use before continuing. For the purposes of this tutorial, identifying configurations with 0.2-nm spacing will suffice, although in the original work a different spacing was used to more thoroughly define the free energy at the energy minimum.
|
Site design and content copyright Justin Lemkul
Problems with the site? Send them to the Webmaster