GROMACS Tutorial
Step Nine: Analysis
|
There are several types of analyses that are particularly useful for membrane protein systems. A brief synopsis will be given here. Several parameters that may be of interest:
Other analyses should be considered, such as secondary structure of the peptide, RMSD, P-N vector orientation, helix tilt, etc. Most of these analyses will require the use of specialized index groups, generated by make_ndx. 1. Deuterium Order Parameters 
For deuterium order parameter analysis, you will need an index group that contains only the carbons along the lipid acyl chains. Each chain must be considered separately! gmx make_ndx -f md_0_1.tpr -o sn1.ndx ... > a C34 > a C36 > a C37 > a C38 ... > a C50 > del 0-21 > q What we have just done is create an index group for the sn-1 chain of DPPC by matching all the atoms in one chain of the lipid with specific atoms names of carbons in the acyl chain. We then deleted all the unnecessary groups (del 0-21). This process should then be repeated for the sn-2 chain (carbons C15, C17-C31 to give "sn2.ndx"). To calculate deuterium order parameters with the normal to the bilayer along the z-axis, use the GROMACS order module: gmx order -s md_0_1.tpr -f md_0_1.xtc -n sn1.ndx -d z -od deuter_sn1.xvg Deuterium order parameters can be helpful in verifying whether or not your membrane entered a gel phase during the simulation. The result of the calculation will look something like this: 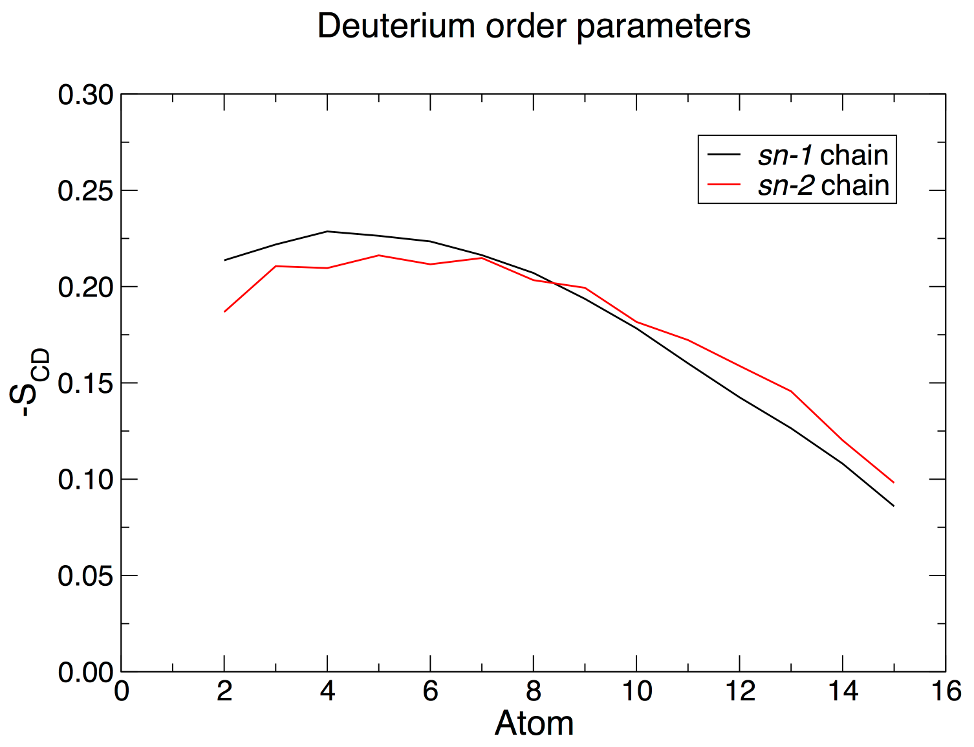
Note that the order program, by default, numbers the carbon atoms in each chain from 1. It is not possible to compute -SCD for terminal carbon atoms, as they lack neighboring atoms from which the local molecular axis is computed. The order program does not know which segment of the acyl chain has been chosen, so it simply numbers the output from 1. The plot above was rendered after re-numbering the output file from 2, reflecting the fact that we have analyzed all applicable carbon atoms in each chain. As such, there are no values for carbon 1 (ester carbon) or 16 (terminal methyl) in each palmitoyl chain. 2. Density of the Membrane One generally sees analysis of membrane density broken down into several different groups - lipid headgroups, acyl chains (sometimes broken down even further to glycerol, methylene, and terminal methyl groups), protein, and solvent. The latter two groups are standard, and do not require any special manipulation. An index group for the lipid headgroups and acyl chains of DPPC can be created by entering the following: gmx make_ndx -f md_0_1.tpr -o density_groups.ndx ... > 13 & a C1 | a C2 | a C3 | a N4 | ... | a O11 > name 22 Headgroups > 13 & a C12 | a C13 | a O14 | a C15 | a O16 | a C32 | a O33 | a C34 | a O35 > name 23 Glycerol_Ester > 13 & ! 22 & ! 23 > name 24 Acyl_Chains > q Now analyze each of these groups separately (note that Protein and SOL are already included in the index file) using the density module: gmx density -s md_0_1.tpr -f md_0_1.xtc -n density_groups.ndx -o dens_headgroups.xvg -d Z Repeat the process for the other groups you wish to analyze, and the result will look similar to the following: 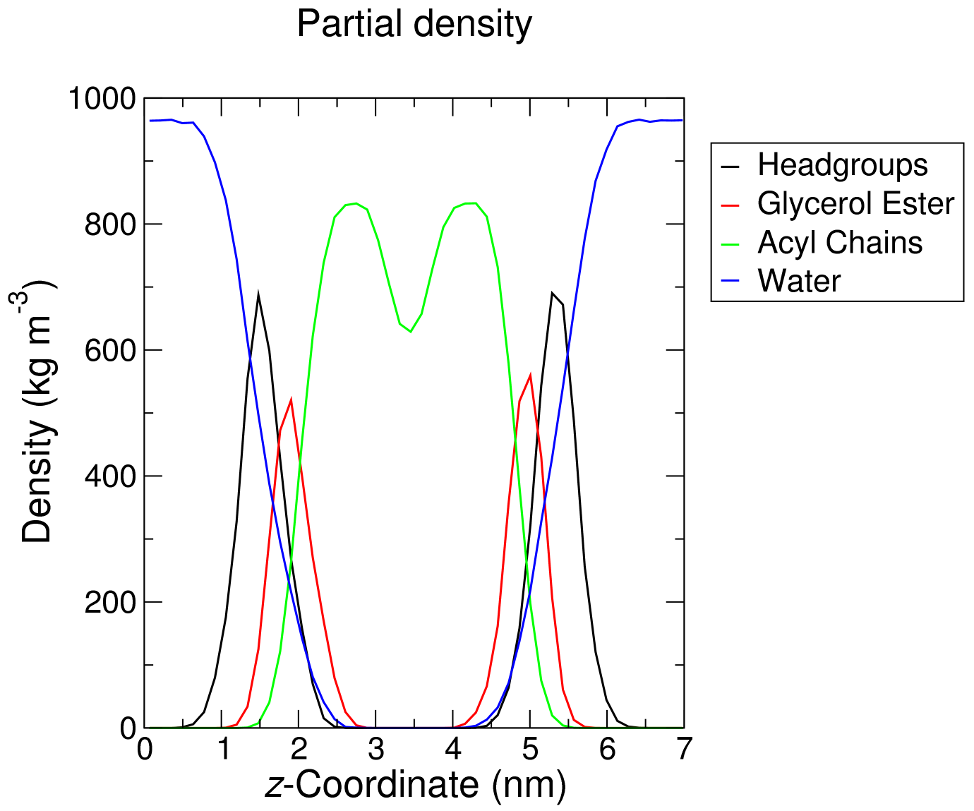
3. Area per Lipid Headgroup and Bilayer Thickness There is no GROMACS tool capable of calculating area per lipid headgroup in the presence of a membrane protein. For a pure membrane, this quantity is simply (Box-X * Box-Y)/(# of lipids per leaflet). The box vectors can be easily extracted from the .edr file using gmx energy. In the case of an embedded protein, such as KALP15, how much space does the protein occupy? We have developed a program that addresses this problem, GridMAT-MD. It calculates area per lipid headgroup, as well as bilayer thickness as a projection across the 2-D bilayer plane (ref). Analyzing area per lipid headgroup is a valuable parameter when verifying that your membrane did not inappropriately enter a gel phase during the simulation. 4. Lateral Diffusion of Lipids GROMACS provides a module called msd to calculate diffusion coefficients. One of its derivative functions is to calculate lateral diffusion (that is, diffusion within a plane, rather than in all three spatial dimensions). This feature is particularly useful for systems containing lipids. To execute msd, you will need to choose one reference atom per lipid, typically P8 of the DPPC headgroup. Make an index group for these atoms: gmx make_ndx -f md_0_1.tpr -o p8.ndx ... > a P8 > q Now, execute msd: gmx msd -s md_0_1.tpr -f md_0_1.xtc -n p8.ndx -lateral z The simulation performed here is insufficient to obtain a reasonable value for the self-diffusion coefficient, e.g.: Fitting from 100 to 900 ps D[ P8] 0.0270 (+/- 0.0267) 1e-5 cm^2/s This value is neither accurate nor precise, but the msd module is illustrated here for its utility in computing important lipid properties.
|
Site design and content copyright Justin Lemkul
Problems with the site? Send them to the Webmaster