GROMACS Tutorial
Step Three: Defining the Unit Cell & Adding Solvent
|
Defining a unit cell for a membrane protein is considerably more complicated than for a protein in water. There are several key steps in building the unit cell:
1. Orient the protein and membrane We have already aligned the KALP peptide using editconf. The bilayer lies in the x-y plane, with the normal along the z-axis. To remove the effects of periodicity, use trjconv: (1) Generate a .tpr file for a DPPC-only system using grompp. You can use any valid .mdp file and a topology corresponding to pure DPPC. An example .mdp file can be found here and such a topology can be found here. Note how simple the topology is. It includes dppc.itp and spc.itp to read parameters for DPPC and water; it's that simple! Run grompp: gmx grompp -f minim.mdp -c dppc128.pdb -p topol_dppc.top -o dppc.tpr You may receive a fatal error like this one, but in this case it is safe to use It is not necessary to run a complete energy minimization procedure on the bilayer, although you can if you want. The .tpr file contains information about bonding and periodicity, so it can, in a sense, be used to reconstruct "broken" molecules. (2) Use trjconv to remove periodicity (select group 0, "System" for output): gmx trjconv -s dppc.tpr -f dppc128.pdb -o dppc128_whole.gro -pbc mol -ur compact Now, take a look at the last line of this .gro file; it corresponds to the x/y/z box vectors of the DPPC unit cell. We need to orient the KALP peptide within this same coordinate frame, and place the center of mass of the peptide at the center of this box: gmx editconf -f KALP-15_processed.gro -o KALP_newbox.gro -c -box 6.41840 6.44350 6.59650 The center of our system now lies at (3.20920, 3.22175, 3.29825), half of each box vector. This is a GROMACS convention. Note that other systems you wish to simulate may not be symmetrical with respect to the membrane, and thus the above command must be modified to something like the following: gmx editconf -f protein.gro -o protein_newbox.gro -box (membrane box vectors) -center x y z In the above command, x y z represents the center of mass such that the protein is properly placed. Placement should be based on experimental knowledge of membrane positioning, or intuition based on the chemical composition of your particular protein. 2. Pack the lipids around the protein The easiest method I have found so far for packing lipids around an embedded protein is the InflateGRO methodology (ref), available here. Please note that I am distributing my own copy of the original version of InflateGRO, not InflateGRO2 available elsewhere from the authors. Download the linked file and rename it "inflategro.pl" to continue. First, concatenate the protein and bilayer structure files: cat KALP_newbox.gro dppc128_whole.gro > system.gro Remove unnecessary lines (the box vectors from the KALP structure, the header information from the DPPC structure) and update the second line of the coordinate file (total number of atoms) accordingly. The authors of the InflateGRO script recommend using a very strong position-restraining force on protein heavy atoms to ensure that the position of the protein does not change during EM. Add a new #ifdef statement to your topology, one that will call a special set of position restraints, such that your topology now contains a section like: ; Include Position restraint file #ifdef POSRES #include "posre.itp" #endif ; Strong position restraints for InflateGRO #ifdef STRONG_POSRES #include "strong_posre.itp" #endif ; Include DPPC chain topology #include "dppc.itp" ; Include water topology #include "gromos53a6_lipid.ff/spc.itp" Now, generate this new position restraint file using genrestr: gmx genrestr -f KALP_newbox.gro -o strong_posre.itp -fc 100000 100000 100000 In the .mdp file used for the minimizations, add a line "define = -DSTRONG_POSRES" to make use of these new position restraints. Then, simply follow the InflateGRO instructions (contained in the script itself), a procedure that is easily scripted. Scale the lipid positions by a factor of 4: perl inflategro.pl system.gro 4 DPPC 14 system_inflated.gro 5 area.dat Since the InflateGRO script requires a very specific order to its command-line arguments, a brief explanation is warranted here. They are:
Note how many lipids were deleted and update the gmx grompp -f minim_inflategro.mdp -c system_inflated.gro -p topol.top -r system_inflated.gro -o system_inflated_em.tpr gmx mdrun -deffnm system_inflated_em The output of mdrun will be "broken" molecules that have all atoms in the central image. As before, reconstruct with trjconv before attempting to use such coordinates with InflateGRO: gmx trjconv -s system_inflated_em.tpr -f system_inflated_em.gro -o tmp.gro -pbc mol mv tmp.gro system_inflated_em.gro At this point, we need to begin packing the lipids around the protein by applying a scaling factor that is < 1. A robust approach is to scale down the lipids by a factor of 0.95, which will require many iterations. The first will be: perl inflategro.pl system_inflated_em.gro 0.95 DPPC 0 system_shrink1.gro 5 area_shrink1.dat Follow this up by another round of EM. During the "shrinking" steps, be sure to keep the InflateGRO cutoff value to 0, or else you will continue to delete lipids! After 26 iterations of scaling down by 0.95, I reached an area per lipid of ~71 Å2, above the experimental value of ~62 Å2. Since the script tends to overestimate the area per lipid, this value is good enough to continue to equilibration. If you wish to automate this process, a Bash script for performing the energy minimization of the inflated system and all subsequent shrinking steps can be found here. Visually, the entire process looks something like this: 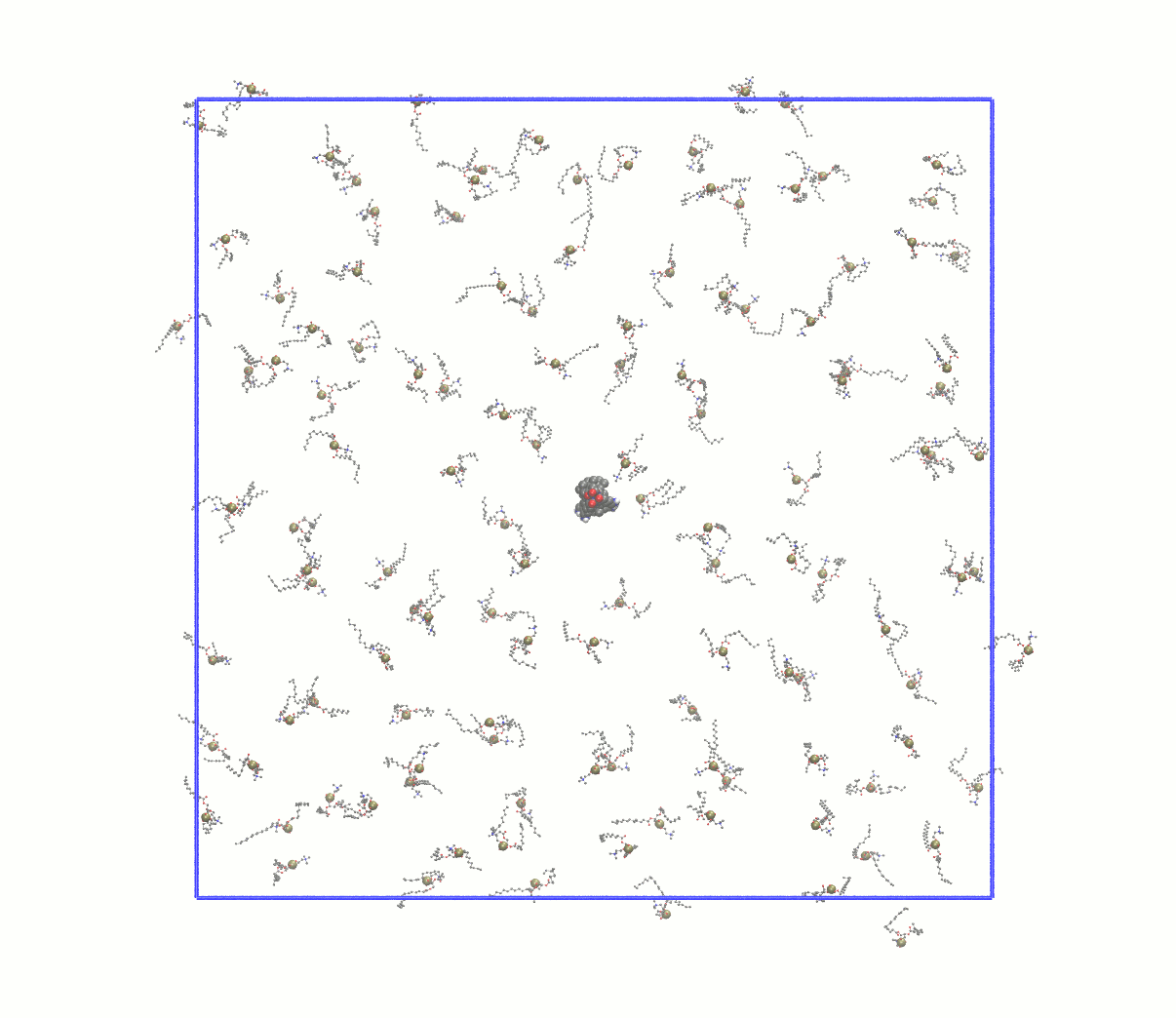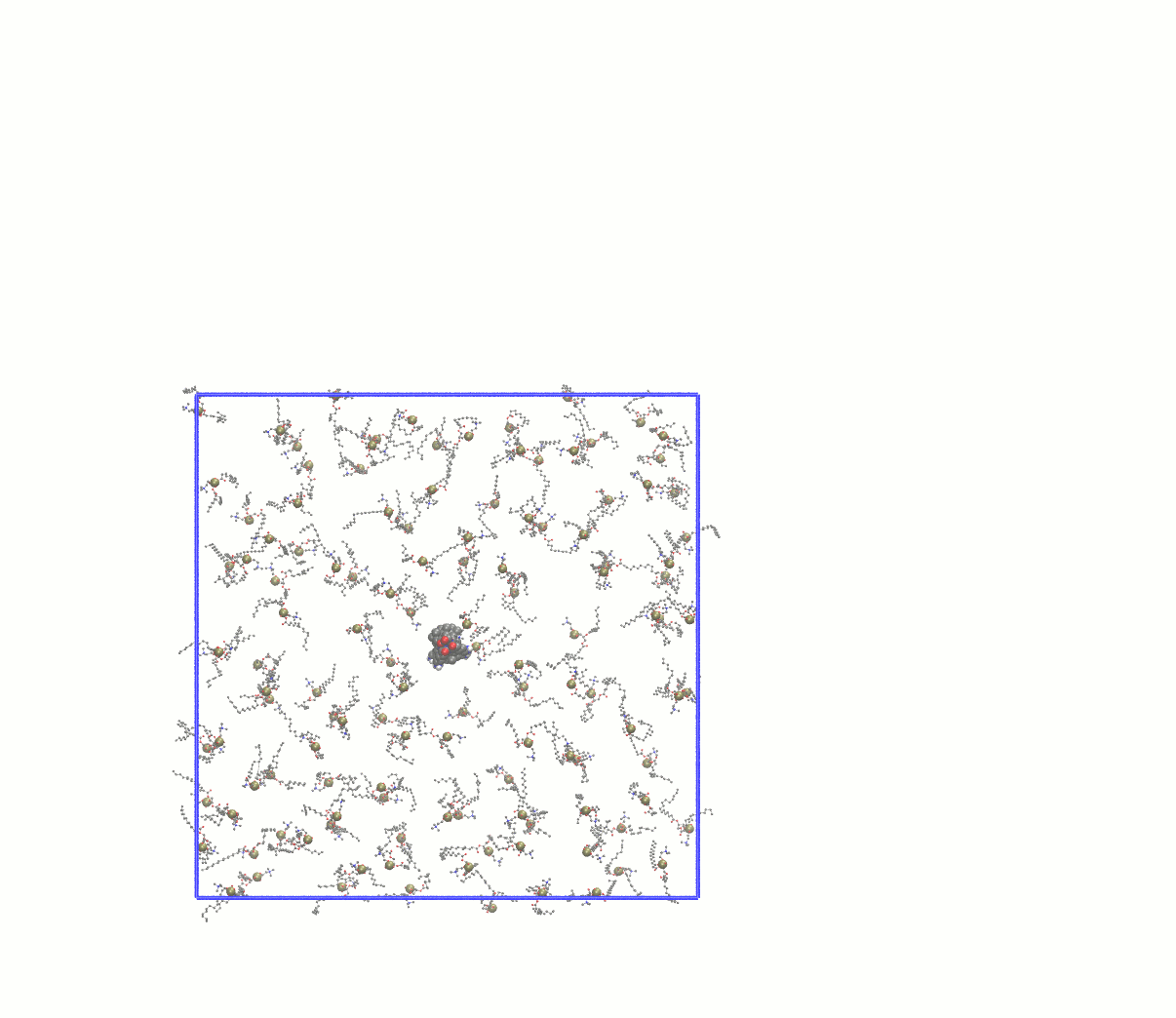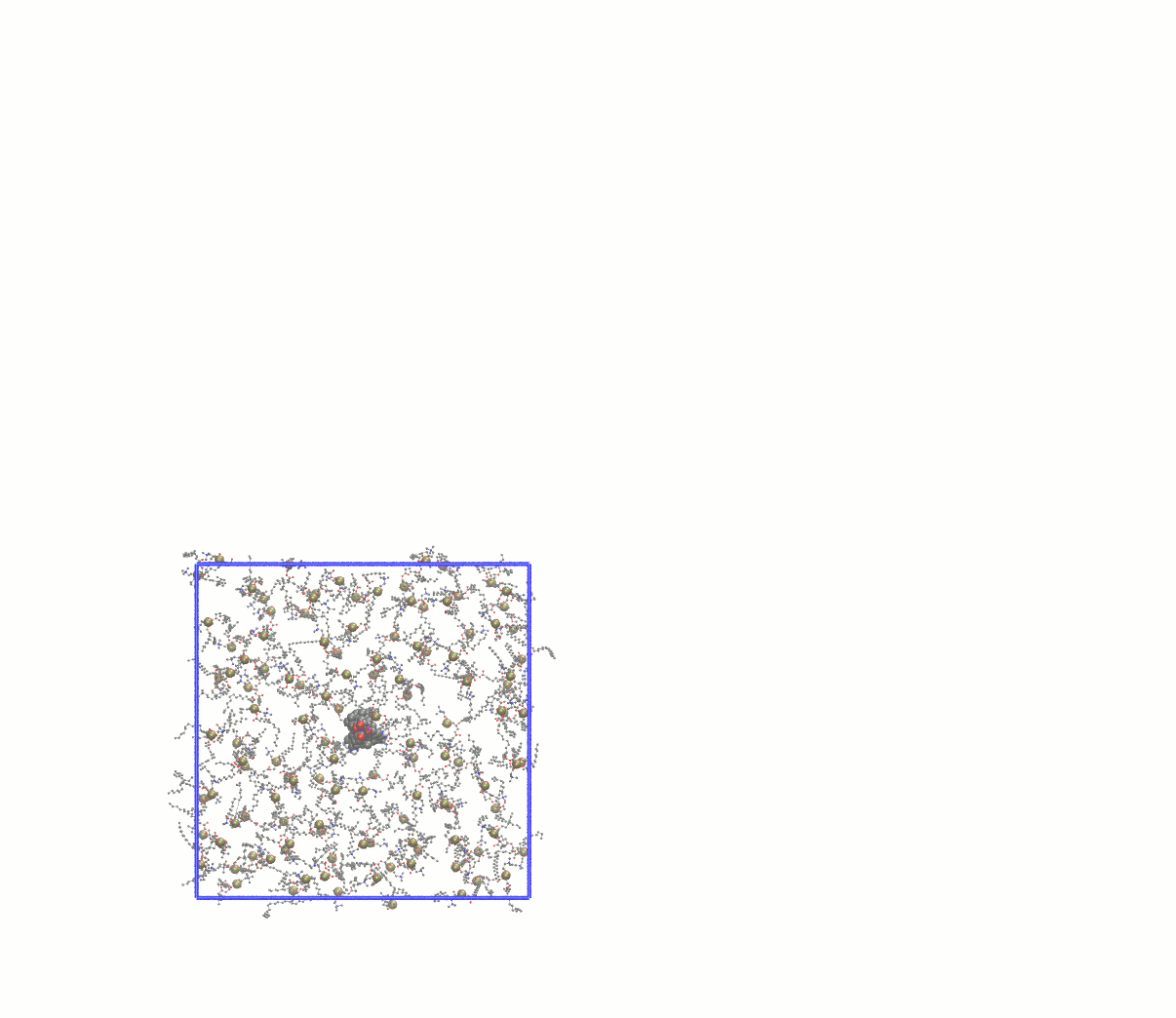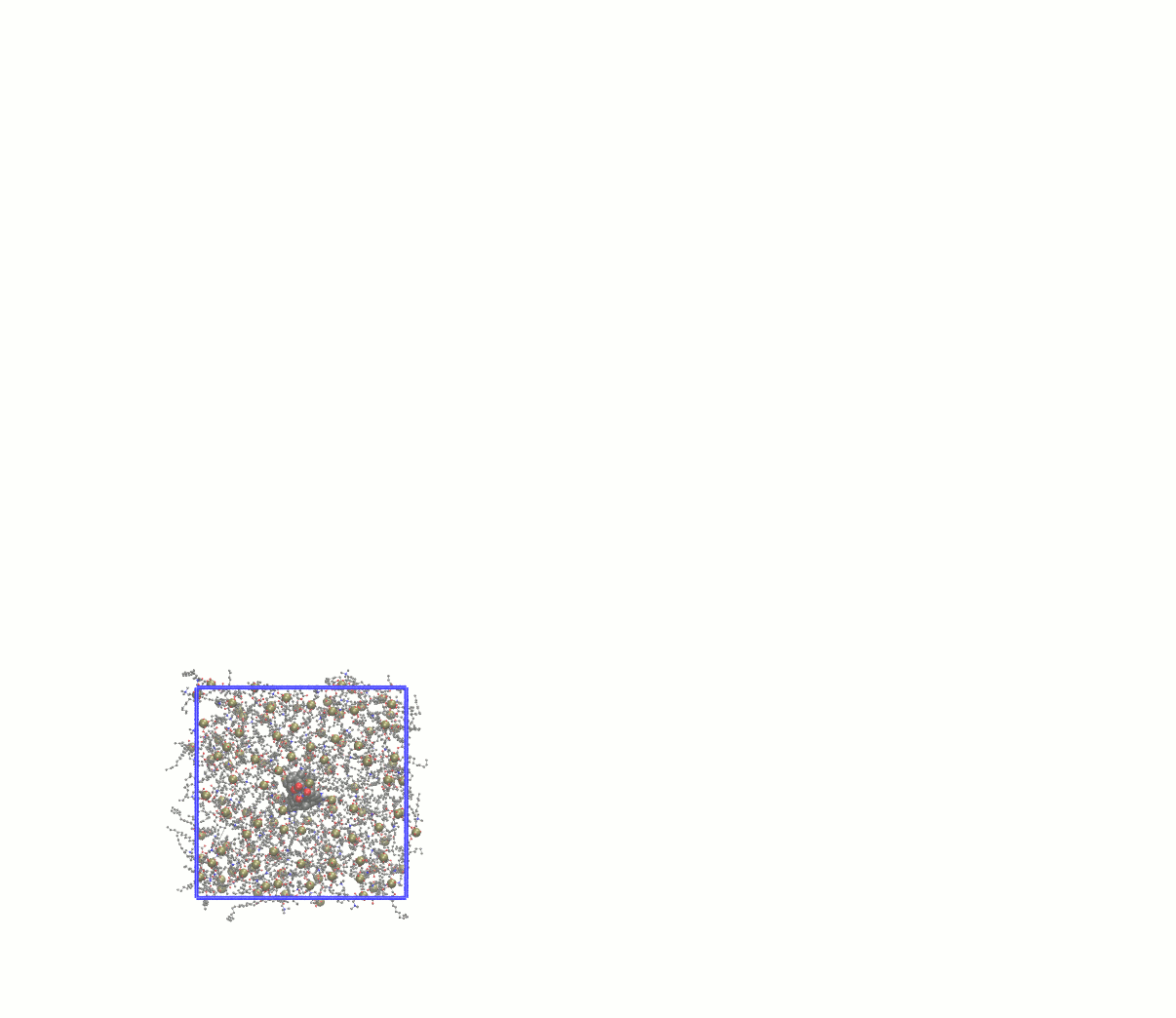
3. Solvate with water Solvating a protein is a trivial task with gmx solvate. Solvating a membrane protein system is not so simple, since the solvate program has a tendency to fill gaps in the lipid acyl chains with water molecules. These water molecules will be deleted later using a custom Perl script. Proceed with solvation as normal: gmx solvate -cp system_shrink26_em.gro -cs spc216.gro -o system_solv.gro -p topol.top After solvating, visualize the structure and you will see many water molecules within the hydrophobic core of the bilayer. Use this Perl script to delete these water molecules: perl water_deletor.pl -in system_solv.gro -out system_solv_fix.gro -ref O33 -middle C50 -nwater 3 The file names passed to -in and -out are the input and output file names, respectively. The -ref flag allows the user to set which atom is set as a "reference" to define the upper and lower boundaries of the membrane. It is wise to set this atom name to one of the atoms in the ester region of the phospholipid rather than a headgroup atom, to prevent excessive dehydration. The -middle flag specifies an atom that is representative of the middle of the bilayer (along the membrane normal, typically the z-axis) so the reference atoms can be divided into upper and lower leaflets, thus establishing the range of z-coordinate values within which water molecules will be deleted. The value passed to -nwater defines how many atoms constitute a water molecule. For SPC, there are only three atoms (OW, HW1, and HW2). The script tells you how many water molecules it deleted and how many water molecules remain in the system. Update the SOL line in topol.top with this updated number of water molecules. You may wish to renumber the coordinate file with gmx genconf, but doing so is not required. |
Site design and content copyright Justin Lemkul
Problems with the site? Send them to the Webmaster